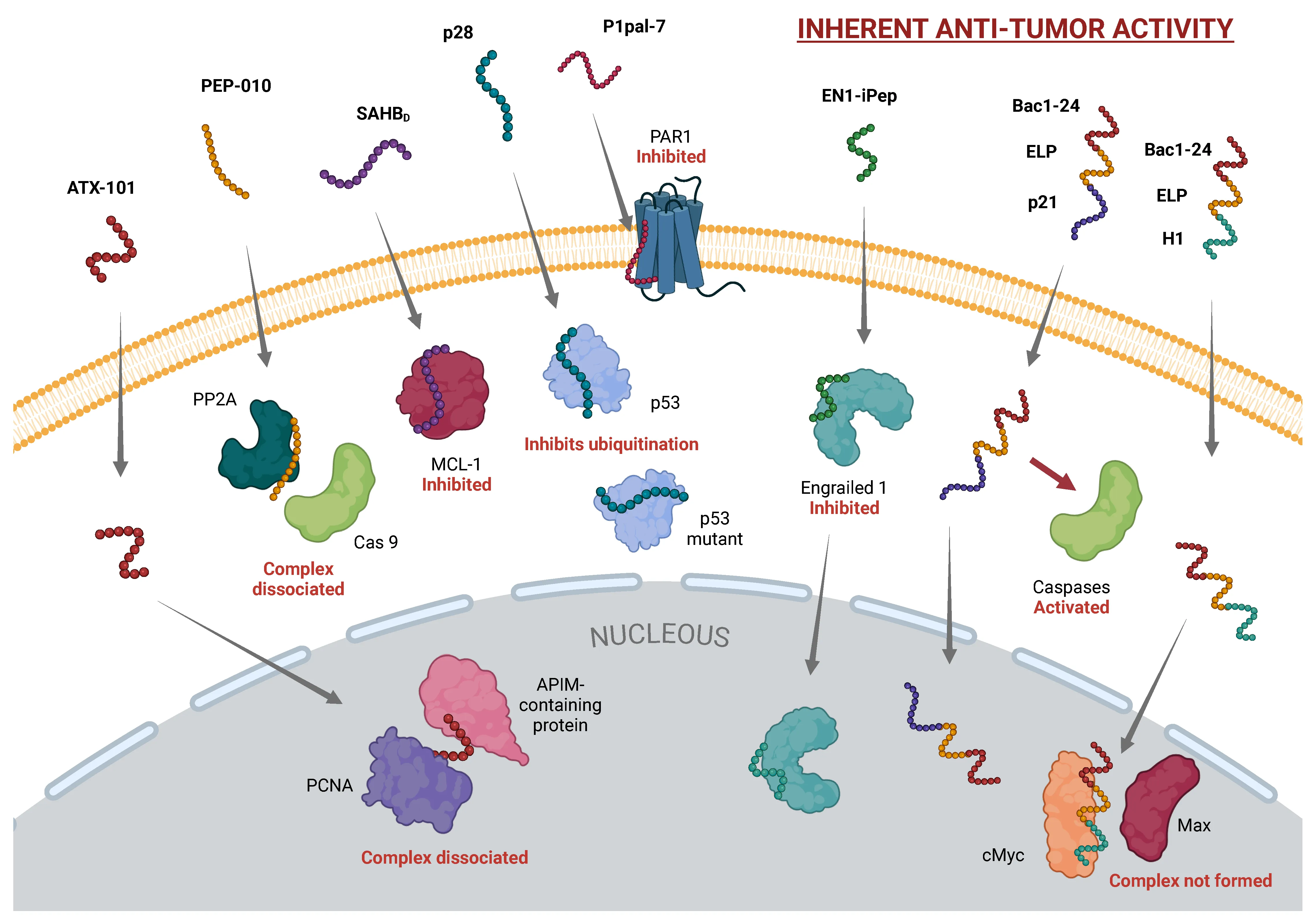
Exploring the Chemical Features and Biomedical Relevance of Cell-Penetrating Peptides
Show abstract Hide abstract
Cell-penetrating peptides (CPPs) are a diverse group of peptides, typically composed of 4 to 40 amino acids, known for their unique ability to transport a wide range of substances—such as small molecules, plasmid DNA, small interfering RNA, proteins, viruses, and nanoparticles—across cellular membranes while preserving the integrity of the cargo. CPPs exhibit passive and non-selective behavior, often requiring functionalization or chemical modification to enhance their specificity and efficacy. The precise mechanisms governing the cellular uptake of CPPs remain ambiguous; however, electrostatic interactions between positively charged amino acids and negatively charged glycosaminoglycans on the membrane, particularly heparan sulfate proteoglycans, are considered the initial crucial step for CPP uptake. Clinical trials have highlighted the potential of CPPs in diagnosing and treating various diseases, including cancer, central nervous system disorders, eye disorders, and diabetes. This review provides a comprehensive overview of CPP classifications, potential applications, transduction mechanisms, and the most relevant algorithms to improve the accuracy and reliability of predictions in CPP development.
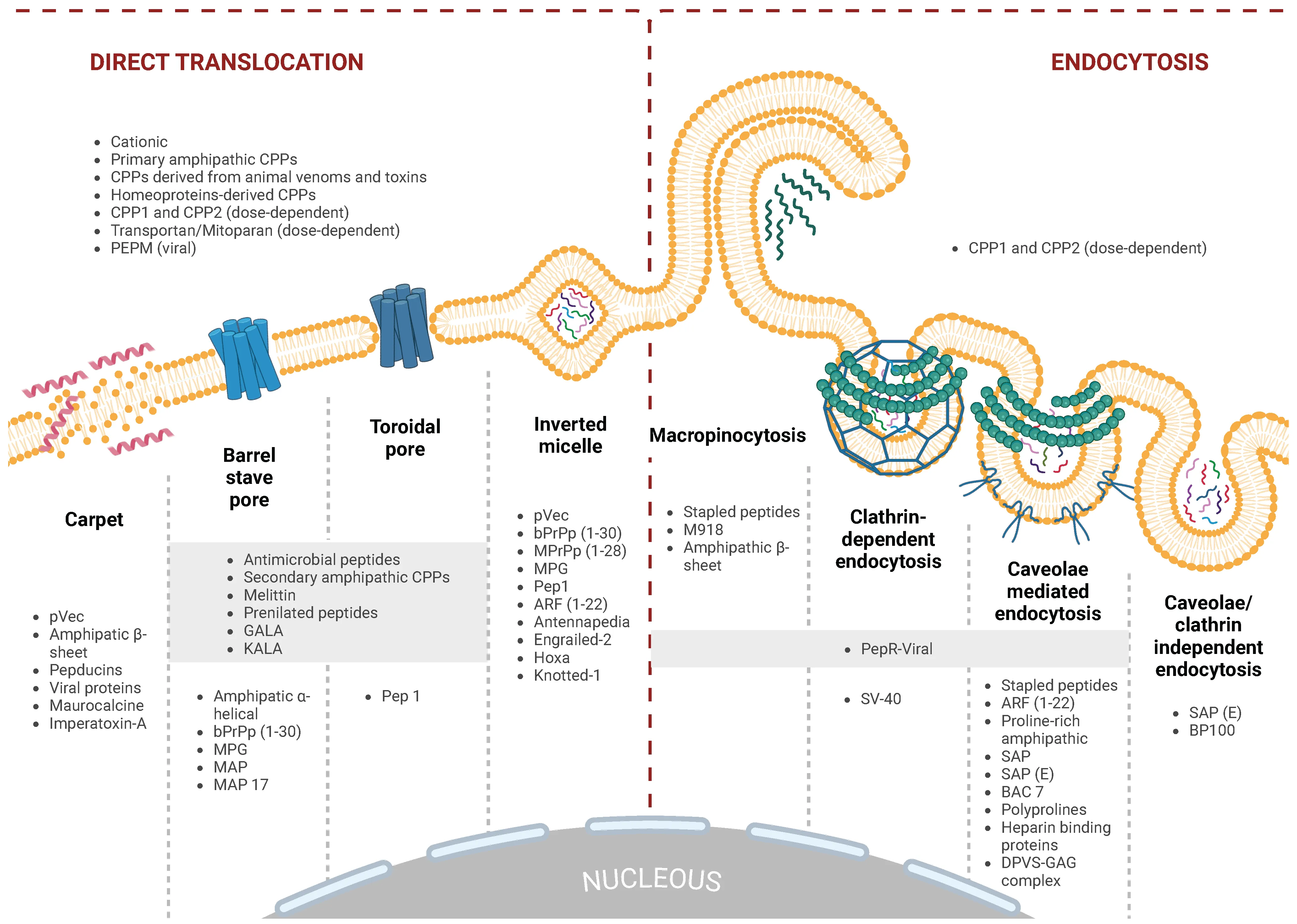
Intracellular entry pathways for CPPs. CPPs utilize two primary mechanisms for cellular entry: energy-dependent endocytosis and energy-independent direct translocation across the lipid bilayer.