Molecular system
With OpenMM from scratch
mport openmm as mm
import openmm.app as app
import openmm.unit as unit
import numpy as np
# Parameters
n_particles = 1
mass = 32.0 * unit.amu
k = 5.0 * unit.kilocalories_per_mole/unit.nanometers**2
# OpenMM topology
topology = app.Topology()
dummy_element = app.Element(0, 'DUM', 'DUM', 0.0 * unit.amu)
chain = topology.addChain('A')
for ii in range(n_particles):
residue = topology.addResidue('DUM', chain)
atom = topology.addAtom(name='DUM', element= dummy_element, residue=residue)
# OpenMM system
system = mm.System()
for ii in range(n_particles):
self.system.addParticle(mass)
force = mm.CustomExternalForce('A*(x^2+y^2+z^2)')
force.addGlobalParameter('A', 0.5*k)
for ii in range(n_particles):
force.addParticle(ii, [])
_ = system.addForce(force)
With this library
This test system is fully documented in HarmonicWell class API. Let’s see an example of how to interact with it:
import numpy as np
from openmm import unit
from matplotlib import pyplot as plt
from uibcdf_systems import HarmonicWell
molecular_system = HarmonicWell(n_particles = 1, mass = 32 * unit.amu,
k=5.0 * unit.kilocalories_per_mole/unit.nanometers**2)
---------------------------------------------------------------------------
ModuleNotFoundError Traceback (most recent call last)
/tmp/ipykernel_3860/1349249507.py in <module>
1 import numpy as np
2 from openmm import unit
----> 3 from matplotlib import pyplot as plt
4
5 from uibcdf_systems import HarmonicWell
ModuleNotFoundError: No module named 'matplotlib'
The potential expression and the value of the parameters are stored in potential:
molecular_system.potential_expression
\[\displaystyle 0.5 k \left(x^{2} + y^{2} + z^{2}\right)\]
molecular_system.parameters
{'n_particles': 1,
'mass': Quantity(value=32, unit=dalton),
'k': Quantity(value=5.0, unit=kilocalorie/(nanometer**2*mole))}
molecular_system.coordinates
Quantity(value=array([[0., 0., 0.]], dtype=float32), unit=nanometer)
molecular_system.topology
<Topology; 1 chains, 1 residues, 1 atoms, 0 bonds>
molecular_system.system
<openmm.openmm.System; proxy of <Swig Object of type 'OpenMM::System *' at 0x7fd269ac9090> >
There is a method to evaluate the potential at a given positions:
molecular_system.evaluate_potential([-1.5, 0.0, 0.0] * unit.nanometers)
Quantity(value=5.625, unit=kilocalorie/mole)
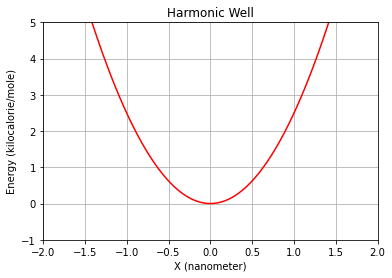
position = np.zeros((200,3), dtype=float) * unit.nanometers
position[:,0] = np.linspace(-5., 5., 200) * unit.nanometers
plt.plot(position[:,0], molecular_system.evaluate_potential(position) , 'r-')
plt.ylim(-1,5)
plt.xlim(-2,2)
plt.grid()
plt.xlabel("X ({})".format(unit.nanometers))
plt.ylabel("Energy ({})".format(unit.kilocalories_per_mole))
plt.title("Harmonic Well")
plt.show()
molecular_system.get_oscillations_time_period()
Quantity(value=7.770948260727904, unit=picosecond)
molecular_system.get_standard_deviation(300.0*unit.kelvin)
Quantity(value=0.34530023967331663, unit=nanometer)
