import openpharmacophore as oph
Protein-ligand complex pharmacophore
We obtain pharmacophores from a md trajectory of er-alpha that consists of three frames
protein = oph.load("../data/eralpha_small.h5")
lig_ids = protein.ligand_ids()
print(lig_ids)
/usr/share/miniconda/envs/test/lib/python3.7/site-packages/mdtraj/core/trajectory.py:438: UserWarning: top= kwargs ignored since this file parser does not support it
warnings.warn('top= kwargs ignored since this file parser does not support it')
[':B']
Note that the trajectory does not contain info of the ligand names, so it’s name appears to be empty. But we can still extract the pharmacophore.
The receptor already contains hydrogens so we don’t need to add any to obtain hydrogen bond acceptor and donor pharmacophoric points
protein.has_hydrogens
<bound method Protein.has_hydrogens of <openpharmacophore.molecular_systems.protein.Protein object at 0x7f9ff79ca110>>
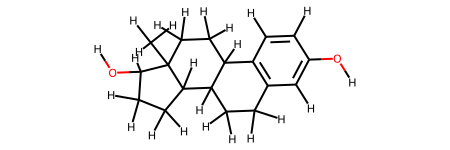
We extract the pharmacophore. We need the smiles of the ligand because its name is not present in the trajectory.
ligand = protein.get_ligand(lig_ids[0], remove_hyd=False)
ligand.fix_bond_order(
smiles="C[C@]12CC[C@@H]3c4ccc(cc4CC[C@H]3[C@@H]1CC[C@@H]2O)O"
)
ligand.draw()
protein.remove_ligand(lig_ids[0])
print(f"Has ligand: {protein.has_ligands}")
Has ligand: <bound method Protein.has_ligands of <openpharmacophore.molecular_systems.protein.Protein object at 0x7f9ff79ca110>>
bsite = oph.ComplexBindingSite(protein, ligand)
pharmacophore = oph.LigandReceptorPharmacophore(bsite, ligand)
pharmacophore.extract(frames=range(3))
for ii, ph in enumerate(pharmacophore):
print(f"Pharmacophore for frame {ii + 1} has {len(ph)} points")
for point in ph:
print(point)
print("\n\n")
Pharmacophore for frame 1 has 5 points
PharmacophoricPoint(feat_type=aromatic ring; center=(30.85, 16.55, 24.52); radius=1.0; direction=(0.01, 0.55, -0.84))
PharmacophoricPoint(feat_type=hb donor; center=(28.34, 17.74, 25.08); radius=1.0; direction=(0.29, 0.57, 0.77))
PharmacophoricPoint(feat_type=hb donor; center=(37.77, 12.08, 23.03); radius=1.0; direction=(-0.32, -0.95, -0.05))
PharmacophoricPoint(feat_type=hb acceptor; center=(37.77, 12.08, 23.03); radius=1.0; direction=(0.65, -0.36, -0.67))
PharmacophoricPoint(feat_type=hydrophobicity; center=(35.22, 12.2, 24.12); radius=1.0)
Pharmacophore for frame 2 has 4 points
PharmacophoricPoint(feat_type=aromatic ring; center=(29.12, 19.02, 22.58); radius=1.0; direction=(0.39, 0.49, -0.78))
PharmacophoricPoint(feat_type=hb donor; center=(26.49, 19.81, 22.45); radius=1.0; direction=(-0.19, 0.93, 0.31))
PharmacophoricPoint(feat_type=hb acceptor; center=(36.18, 15.29, 24.48); radius=1.0; direction=(0.65, -0.69, -0.31))
PharmacophoricPoint(feat_type=hydrophobicity; center=(33.49, 15.42, 24.81); radius=1.0)
Pharmacophore for frame 3 has 4 points
PharmacophoricPoint(feat_type=aromatic ring; center=(24.07, 22.89, 23.78); radius=1.0; direction=(-0.08, 0.42, -0.9))
PharmacophoricPoint(feat_type=hb acceptor; center=(32.19, 20.78, 24.97); radius=1.0; direction=(0.9, -0.39, -0.21))
PharmacophoricPoint(feat_type=hydrophobicity; center=(30.01, 20.08, 23.73); radius=1.0)
PharmacophoricPoint(feat_type=hydrophobicity; center=(29.32, 20.32, 25.93); radius=1.0)
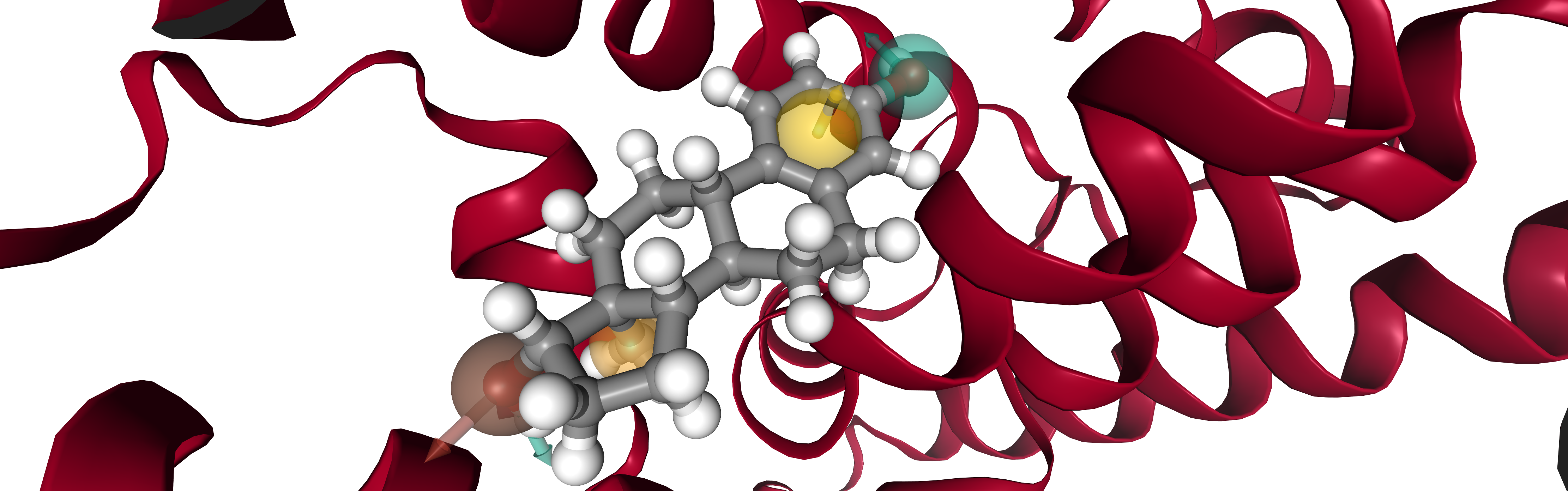
viewer = oph.Viewer()
viewer.add_components([protein, ligand, pharmacophore])
viewer.show(struct=0)
viewer.show(struct=2)
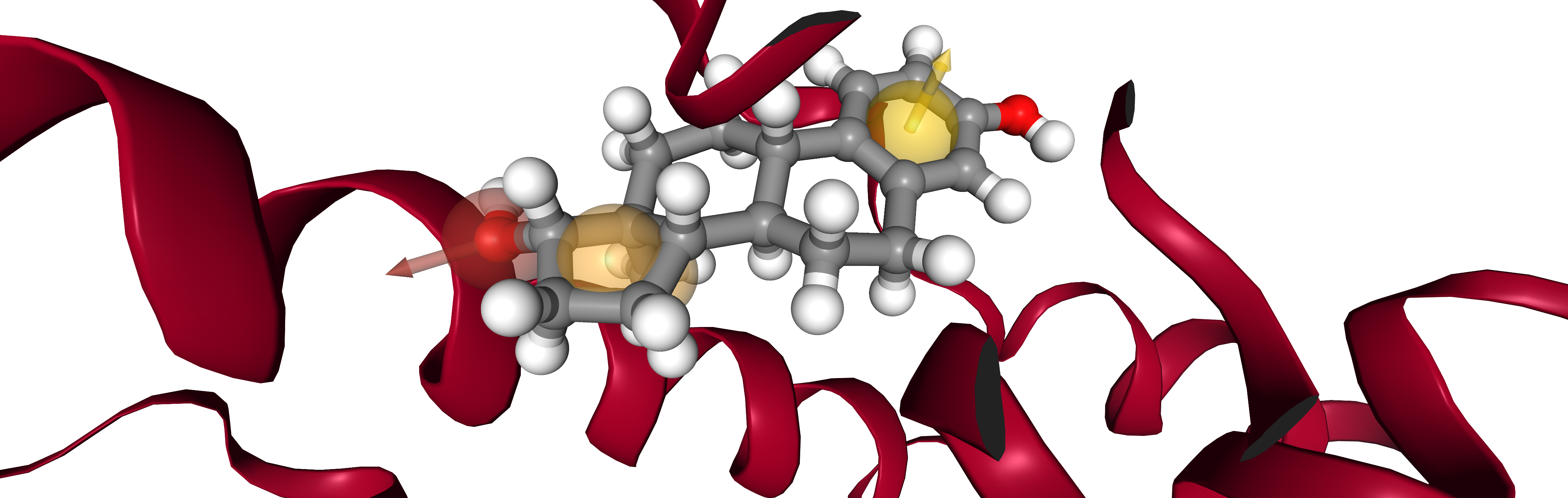
Note:
viewer.show() displays an interactive widget. For simplicity an image is presented in the documentation.
