Human Rhinovirus Serotype
import os
import openpharmacophore as oph
import pyunitwizard as puw
from openpharmacophore.pharmacophore.align import align_pharmacophores
Load and visualize protein-ligand complexes
pdbs_path = "../data/hrv/"
def load_proteins():
prots = [
oph.load(os.path.join(pdbs_path, "1c8m.pdb")),
oph.load(os.path.join(pdbs_path, "1ncr.pdb")),
oph.load(os.path.join(pdbs_path, "1nd3.pdb")),
]
print(prots[0].ligand_ids())
print(prots[1].ligand_ids())
print(prots[2].ligand_ids())
return prots
proteins = load_proteins()
['W11:E']
['MYR:F', 'W11:E']
['W11:E']
viewer = oph.Viewer()
viewer.add_components(proteins)
viewer.show()
Extracting ligands
We extract the ligands with code W11 that corresponds to pleconaril.
lig_id = proteins[0].ligand_ids()[0]
smiles = oph.smiles_from_pdb_id(lig_id)
smiles
'Cc1cc(cc(c1OCCCc2cc(no2)C)C)c3nc(on3)C(F)(F)F'
def load_ligands():
lig_list = [p.get_ligand(lig_id) for p in proteins]
for lig in lig_list:
lig.fix_bond_order(smiles=smiles)
lig.add_hydrogens()
return lig_list
ligands = load_ligands()
oph.draw_ligands(ligands, n_per_row=3)
Preparing proteins
for prot in proteins:
prot.extract_chain("A")
prot.add_hydrogens()
assert not prot.has_ligands()
assert not prot.has_solvent_or_ions()
viewer = oph.Viewer()
viewer.add_components(proteins + ligands)
viewer.show()
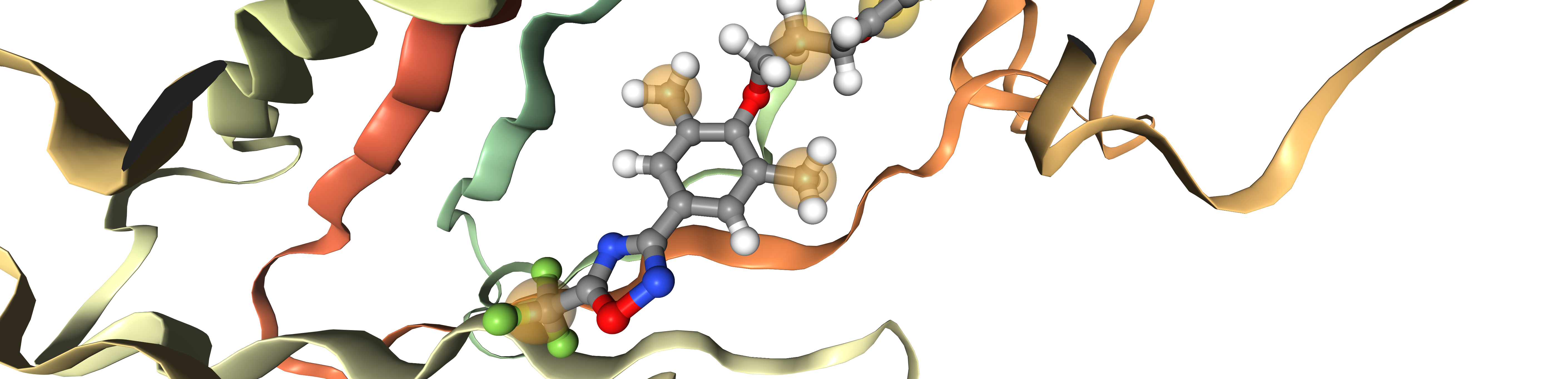
Obtaining binding sites
binding_sites = []
for ii in range(len(proteins)):
binding_sites.append(oph.ComplexBindingSite(proteins[ii], ligands[ii]))
Extracting pharmacophores
pharmacophores = []
for ii in range(len(binding_sites)):
lrp = oph.LigandReceptorPharmacophore(binding_sites[ii], ligands[ii])
lrp.extract()
print(f"\nPharmacophore {ii + 1}")
for pnt in lrp[0]:
print(pnt)
pharmacophores.append(lrp)
Pharmacophore 1
PharmacophoricPoint(feat_type=hydrophobicity; center=(48.89, -4.4, 121.79); radius=1.0)
PharmacophoricPoint(feat_type=hydrophobicity; center=(40.93, 3.43, 122.23); radius=1.0)
PharmacophoricPoint(feat_type=hydrophobicity; center=(45.04, 3.15, 123.78); radius=1.0)
PharmacophoricPoint(feat_type=hydrophobicity; center=(41.87, -0.79, 122.69); radius=1.0)
Pharmacophore 2
PharmacophoricPoint(feat_type=aromatic ring; center=(38.03, 4.36, 123.74); radius=1.0; direction=(-0.67, -0.05, -0.74))
PharmacophoricPoint(feat_type=hydrophobicity; center=(49.65, -4.36, 122.0); radius=1.0)
PharmacophoricPoint(feat_type=hydrophobicity; center=(41.14, 2.96, 122.61); radius=1.0)
PharmacophoricPoint(feat_type=hydrophobicity; center=(45.46, 2.01, 123.42); radius=1.0)
PharmacophoricPoint(feat_type=hydrophobicity; center=(41.37, -1.11, 122.68); radius=1.0)
Pharmacophore 3
PharmacophoricPoint(feat_type=aromatic ring; center=(37.82, 4.36, 123.82); radius=1.0; direction=(-0.64, -0.06, -0.77))
PharmacophoricPoint(feat_type=hydrophobicity; center=(49.44, -4.36, 122.09); radius=1.0)
PharmacophoricPoint(feat_type=hydrophobicity; center=(40.93, 2.96, 122.69); radius=1.0)
PharmacophoricPoint(feat_type=hydrophobicity; center=(45.25, 2.0, 123.51); radius=1.0)
PharmacophoricPoint(feat_type=hydrophobicity; center=(41.16, -1.11, 122.76); radius=1.0)
idx = 2
viewer = oph.Viewer()
viewer.add_components([
proteins[idx],
ligands[idx],
pharmacophores[idx],
])
viewer.show()
Finding a common pharmacophore
To find a common pharmacophore we align the pharmacophores with one another. We’ll choose the one with lowest rmsd as the consensus pharmacophore
pharma_mat = [
puw.get_value(p[0].to_matrix()) for p in pharmacophores
]
hydrophobics = [
pharma_mat[0], pharma_mat[1][1:, :], pharma_mat[2][1:, :]
]
Align hydrophobic features
def align_and_score(matrices):
rmsd_list = []
for ii in range(len(matrices)):
avg_rmsd = 0
for jj in range(len(matrices)):
if ii == jj:
continue
rmsd, _ = align_pharmacophores(matrices[ii], matrices[jj])
avg_rmsd += rmsd
avg_rmsd /= (len(matrices) - 1)
rmsd_list.append(avg_rmsd)
return rmsd_list
rmsd_list = align_and_score(hydrophobics)
rmsd_list
[0.671464357705515, 0.3367555679305882, 0.33734245356241305]
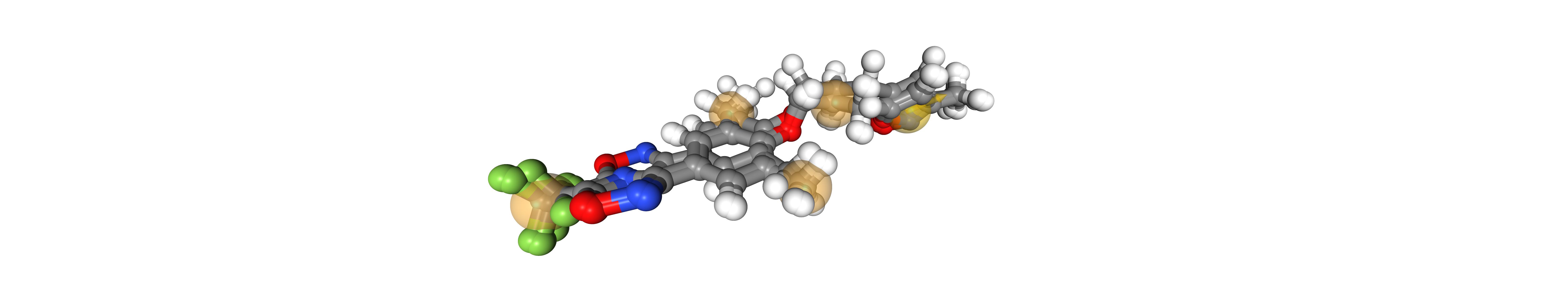
Taking the second pharmacophore as the alignment reference gives the lowest RMSD. So we take this as our common pharmacophore
viewer = oph.Viewer()
viewer.add_components(ligands + [pharmacophores[2]])
viewer.show()
Align hydrophobic features and aromatic ring
rmsd_list = align_and_score(pharma_mat[1:])
rmsd_list
[0.0025020000021948063, 0.0025020000021948063]
viewer = oph.Viewer()
viewer.add_components(ligands + [pharmacophores[1]])
viewer.show()