Add constant force#
import molsysmt as msm
import openmm as mm
from openmm import unit
from openmm import app
from tqdm import tqdm
import numpy as np
from matplotlib import pyplot as plt
molecular_system = msm.systems['Trp-Cage']['1l2y.h5msm']
topology = msm.convert(molecular_system, to_form='openmm.Topology')
positions = msm.get(molecular_system, element='atom', structure_indices=0, coordinates=True)
positions = msm.pyunitwizard.convert(positions[0], to_form='openmm.unit')
forcefield = app.ForceField("amber14-all.xml", "amber14/tip3p.xml")
system = forcefield.createSystem(topology, nonbondedMethod=app.NoCutoff, constraints=app.HBonds)
temperature = 300*unit.kelvin
integration_timestep = 2.0*unit.femtoseconds
saving_interval = 1.00*unit.picoseconds
logging_interval = 100.00*unit.picoseconds
simulation_time = 500.*unit.picoseconds
saving_steps = int(saving_interval/integration_timestep)
logging_steps = int(logging_interval/integration_timestep)
md_steps = int(simulation_time/integration_timestep)
friction = 5.0/unit.picoseconds
integrator = mm.LangevinIntegrator(temperature, friction, integration_timestep)
platform = mm.Platform.getPlatformByName('OpenCL')
simulation = app.Simulation(topology, system, integrator, platform)
simulation.context.setPositions(positions)
msm.get(simulation, n_groups=True)
20
msm.info(simulation, element='atom', selection='group_index==0 and atom_type=="C"')
| index | id | name | type | group index | group id | group name | group type | component index | chain index | molecule index | molecule type | entity index | entity name |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | CA | C | 0 | 1 | ASN | amino acid | 0 | 0 | 0 | peptide | 0 | None |
| 2 | 3 | C | C | 0 | 1 | ASN | amino acid | 0 | 0 | 0 | peptide | 0 | None |
| 4 | 5 | CB | C | 0 | 1 | ASN | amino acid | 0 | 0 | 0 | peptide | 0 | None |
| 5 | 6 | CG | C | 0 | 1 | ASN | amino acid | 0 | 0 | 0 | peptide | 0 | None |
msm.info(simulation, element='atom', selection='group_index==19 and atom_type=="C"')
| index | id | name | type | group index | group id | group name | group type | component index | chain index | molecule index | molecule type | entity index | entity name |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 293 | 294 | CA | C | 19 | 20 | SER | amino acid | 0 | 0 | 0 | peptide | 0 | None |
| 294 | 295 | C | C | 19 | 20 | SER | amino acid | 0 | 0 | 0 | peptide | 0 | None |
| 296 | 297 | CB | C | 19 | 20 | SER | amino acid | 0 | 0 | 0 | peptide | 0 | None |
msm.thirds.openmm.forces.pin_atoms(simulation, selection='group_index==0 and atom_name=="CA"')
5
msm.thirds.openmm.forces.add_constant_force(simulation, selection='group_index==19 and atom_name=="CA"',
force = '[500,0,0] kilojoules/(mole*nanometer)')
6
reporter_tqdm = msm.thirds.openmm.reporters.TQDMReporter(logging_steps, md_steps, temperature=False)
simulation.reporters.append(reporter_tqdm)
reporter_trajectory_dict = msm.thirds.openmm.reporters.TrajectoryDictReporter(saving_steps, time=True, coordinates=True)
simulation.reporters.append(reporter_trajectory_dict)
simulation.step(md_steps)
Potential energy: -1714.79 kJ/mol ± 204.61 kJ/mol
Execution time: 0 days, 0 hours, 0 minutes, and 14.77 seconds (2924.449 ns/day).
trajectory_dict = reporter_trajectory_dict.finalize()
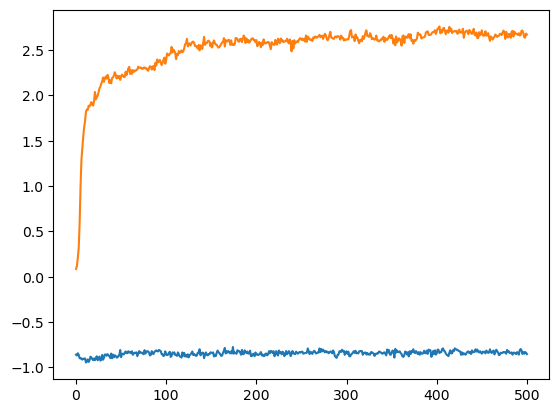
index_CA1, index_CA20 = msm.get(topology, element='atom', selection='group_index in [0,19] and atom_name=="CA"', atom_index=True)
plt.plot(trajectory_dict['time'], trajectory_dict['coordinates'][:,index_CA1,0])
plt.plot(trajectory_dict['time'], trajectory_dict['coordinates'][:,index_CA20,0])
plt.show()
msm.view([topology, trajectory_dict])